



.jpg)
Last updated: November 16, 2025
Why risk-based TMF frameworks are now essential
The U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) now require sponsors to implement risk-based approaches to clinical trial quality management.
As trials have grown more complex with diverse patient populations, intricate protocols, and advancing technologies, traditional uniform documentation processes have become inefficient. Regulatory agencies focus on improving trial quality, participant safety, and data integrity through risk-based methodologies.
A risk-based TMF framework identifies, assesses, mitigates, and monitors risks in trial documentation activities. Rather than applying uniform procedures to all documents, this framework allocates quality control resources based on the level of risk associated with specific artifacts, processes, and data.
This guide walks you through Montrium's 5-step approach to implementing a risk-based TMF framework.
What is a risk-based TMF framework?
A risk-based approach to Trial Master File (TMF) management involves identifying, assessing, mitigating, and monitoring risks inherent in clinical trial documentation activities. Rather than applying uniform procedures to all documents, the framework allocates resources and quality control interventions based on the level of risk associated with specific artifacts, processes, and data.
Montrium's risk-based TMF framework includes five key steps:
- Understand the existing risks in your TMF process: Evaluate process-level risks through a TMF Risk Management Plan and Risk Log
- Assess core vs recommended documents: Distinguish essential documents from supplementary artifacts
- Evaluate impact on patient safety and data integrity: Classify artifacts by their potential impact
- Analyze non-compliance with TMF principles: Calculate risk scores based on completeness, quality, and timeliness
- Consider additional risk factors: Account for document lifecycle, translations, SOPs, and quality events
This framework enables sponsors to focus quality control efforts on highest-risk areas while maintaining inspection readiness.
The steady emergence of risk-based TMF management
Risk-based TMF management is not a concept that appeared out of thin air. This approach is the culmination of years of procedural and technological evolution within the clinical research industry. It is a milestone reflected in the January 2025 adoption of ICH E6(R3), which mandates risk-based approaches to quality management across all clinical trial activities.
Take, for example, this fundamental paragraph from ICH-GCP E6(R3):
“The sponsor should implement an appropriate system to manage quality throughout all stages of the trial process. Quality management includes the design and implementation of efficient clinical trial protocols including tools and procedures for trial conduct (including for data collection and management) in order to support participant’s rights, safety and well-being and the reliability of trial results.
The sponsor should adopt a proportionate and risk-based approach to quality management, which involves incorporating quality into the design of the clinical trial (i.e. quality by design) and identifying those factors that are likely to have a meaningful impact on participant’s rights, safety and well-being and the reliability of the results (i.e., critical to quality factors as described in ICH E8(R1)). The sponsor should describe the quality management approach implemented in the trial in the clinical trial report.” ICH-GCP R3 Guidelines (ICH E6(R3) 3.10) Quality Management
Indeed, the regulatory landscape is clear: companies must implement and develop a risk-based approach to ensure quality in clinical trials, but how? The regulations and official guidance do not provide the clear, detailed steps and practical actions required for implementing a risk-based approach, especially when it comes to the TMF.
The question is not whether to develop a risk-based TMF framework, but how to implement one effectively.
Developing a risk-based approach to TMF for inspection readiness
When evaluating risks associated with the Trial Master File (TMF), the biggest risk that we usually identify is failing an inspection due to insufficient or incorrect information within the TMF.
Regulatory inspection data confirms this risk. A peer-reviewed analysis of FDA and European Medicines Agency (EMA) GCP inspections conducted between 2009 and 2015 found that documentation deficiencies, including TMF-related findings, showed a 70% concordance rate between the two agencies. For EMA inspections specifically, documentation deficiencies represented the most common finding category for both clinical investigator and sponsor/CRO inspections, underscoring the regulatory focus on TMF completeness and quality.
So, how can we ensure that our TMF is in compliance with what inspectors are asking for?
If the TMF is not complete, the documentation is not accurate, or the documents have not been filed in a timely manner, there is a concrete risk of receiving critical or major findings that must be fixed immediately. In more serious cases, we may even fail the inspection.
There are incredibly large numbers of documents and stakeholders (sponsors, CROs, sites, vendors, etc.) involved in the production of TMF artifacts. Having so many moving parts means that the goal of being in an inspection-ready state at all times seems unrealistic, with many sponsors struggling to achieve it today.
Regulatory authorities are aware of this fact, which is why they encourage a “proportionate approach.” A proportionate approach allows sponsors to develop a risk-based approach (RBA) to optimize time and resources when managing the quality of the TMF.
Applying an RBA to the TMF allows sponsors to focus on what really matters in the story of the clinical trial and what can really support the sponsor in demonstrating data integrity and patients’ well-being during inspections.
Common circumstances in TMF management from which quality issues arise include:
- A core document was never collected
- Documentation was filed months after the date of creation
- Poor quality artifacts with regards to ALCOA++
- Misalignment with trial plans
- Too many days to close a quality query
- Duplicate artifacts
- Lack of consistency
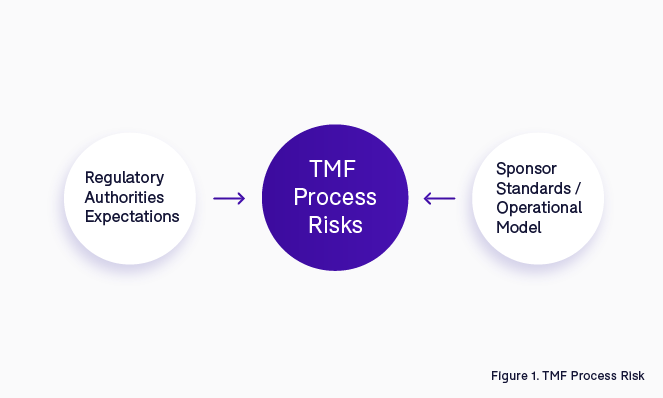
In order to avoid or mitigate these types of issues, it is important to understand in advance what could be considered a potential regulatory finding within the TMF management process. Combining regulatory authorities’ expectations with internal company knowledge and operational models is the starting point to identify what the risks are in the TMF process (Figure 1).
Example: Process Consistency
Let us look at an example to clarify what we mean. Process consistency is a common expectation that inspectors look for. Inspectors usually do not have much time to open every single document within the TMF to understand its content; instead, the title must speak to them and help them to identify the content.
For example, in the case of four different CVs from site staff, they would not want to see four different ways of naming the same type of document:
- PerryMason_CV_2023;
- MJ_CV_Nov2021;
- CurriculumBatman_21;
- 210305_ABC_CV.
Inspectors are looking for consistency. They are looking for clear rules applied to each document that will simplify their job and will be a sign of accuracy and quality in terms of the sponsor’s documentation management process.
The steps of the TMF process that contain risks must be assessed and captured from the beginning of the trial to ensure higher quality. Once we have defined the risks at TMF process level, it is important to monitor those risks to avoid unnecessary risk escalation and negative effects on the TMF’s inspection-ready state.
The activity of checking and constantly monitoring is done through regular TMF quality control (QC). Only by checking the TMF with a pre-defined frequency can we identify issues and gaps in advance, and more importantly, have time to fix them before an inspection occurs.
Setting the foundations for a risk-based approach
How can TMF teams check thousands of documents effectively when frequently it is just one resource (namely trial CTA) who is responsible for the task?
The answer is simple: applying a risk-based approach. However, the underlying question still remains, how can we do that? If we think about all the artifacts that are contained in a TMF, we can certainly affirm that not all of them have the same weight in terms of risk to patient safety and data integrity.
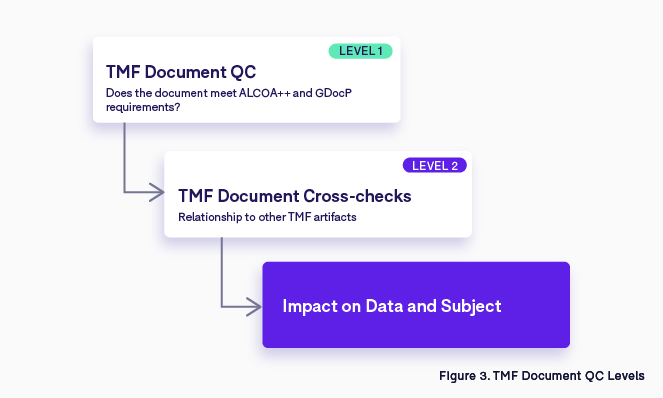
Some are more critical than others; if you look at the CDISC TMF Reference Model, there is a clear distinction between what is core and what is recommended. In ICH-GCP they refer to Essential Documents, making a reference only to those documents that enable the reconstruction of the clinical trial (Figure 3).

Because of the diversity in the nature of these documents, some of them require more attention than others. Not every document is essential to tell the story; for example, a Protocol Amendment has much more value than a Site Newsletter.
Example: Delegation log risk
Let’s look at an example: think about a Delegation Log, a document issued at site level which contains the list of people authorized by a Principal Investigator to work in a clinical trial. Due to the data contained in this document, the quality check will be done at two different levels:
- The first level check is to verify that the document meets the requirements of ALCOA++ and GDocP (this level of quality has to be met for every single artifact in the TMF).
- The second level of QC tends to focus on the relationships with other TMF artifacts—for each person listed on the delegation log, other essential documents must be collected and filed such as the CV, the GCP training certificate, the medical license, the protocol training certificate, etc.
The Delegation Log has a clear and important impact on patient safety and data integrity (driven by principles of ICH-GCP), which is why it would be classified as high risk (Figure 4) from a RBA QC perspective. Think about the situation whereby an inspector finds a name on a visit record that does not appear in the Delegation Log, and furthermore, the individual's CV is also missing. The immediate thought could be that someone who is not authorized has given treatment to a trial subject. Without the correct documentation, it would be challenging to demonstrate that person is part of the site staff.
Distinguishing types of risks
When we start to develop a risk-based approach to TMF management, the initial distinction that we need to establish is between the different types of risks. In our road towards an inspection-ready TMF, we must tackle not only risks associated with the TMF process, but also risks associated with the TMF artifacts themselves (Figure 5).
The starting point of taking a risk-based approach is the initial assessment of risk, which means identifying the potential risks that can afflict the quality of our final deliverable. In our case, this would be the trial TMF.
Making a clear distinction between what is process-related and what is artifact-related will help to develop a better strategy for TMF risk management, including the ability to identify preventable risks that can be mitigated successfully through preventative actions.

Establishing and assessing risk in the TMF
Step 1: Understand the existing risks in your TMF process
If people inside the organization are not trained on TMF requirements, TMF SOPs, and trial plans, do you think that the story of your clinical trial will be complete at the end of the study?
The different steps of the TMF process can have an impact on data integrity and subject safety, and it is important that we evaluate each of them for potential risks. For example, if you do not have proper rules to manage user access to the eTMF, there could be a potential risk of accidental unblinding.
In order to manage this part of the risk-based approach process, there are two key guidance and tracking documents that need to be developed:
- A TMF Risk Management Plan
- A TMF Risk Management Log
Defining risks in TMF management involves recognizing events or conditions that, upon occurrence, may impact a trial's objectives positively or negatively. The comprehensive process of risk management encompasses the identification, assessment, response formulation, continuous monitoring, and reporting of risks across all trial activities based on trial design.
Once the overall trial risk assessment has been completed, we need to evaluate the identified risk of impact on the TMF and TMF management process. These TMF-specific risks should then be logged in the TMF Risk Management Log and potentially added to the overall trial risk management log when appropriate.
TMF risk management plan
The TMF Risk Management Plan will support your team when they identify the potential risks in the TMF process in collaboration with all relevant TMF stakeholders. Although regulatory authorities mandate that the trial's eTMF be inspection ready at all times, inherent risks exist within the TMF process steps.
This Risk Management Plan for the TMF articulates the approach to identifying, analyzing, and managing risks associated with the trial's TMF management and with the overall trial risks. It delineates the methodologies employed for performing, documenting, and overseeing TMF risk management activities throughout the trial's lifecycle.
How to use the TMF Risk Management Plan:
- Identification of TMF Risks: In addition to the standard TMF management risks that can occur with every trial, we also need to identify potential trial-specific risks associated with TMF management that could occur based on our evaluation of the trial risk log. This includes risks related to processes, personnel, and technology.
- TMF Risk Assessment: Once risks are identified, refer to the risk analysis section in the plan. Assess and classify the potential impact and likelihood of each identified risk. This step aids in prioritizing risks for effective management.
- TMF Risk Score: Define the trial specific methodology and parameters for the calculation of risk scores for the various dimensions of the trial and the types of risks based on issues observed within the trial. Risk scoring may also use historical data from other studies.
- TMF Risk Management Strategy: Clearly define the risk-based oversight and TMF management strategies and activities for artifacts based on the various risk factors identified.
- TMF Risk Monitoring, Controlling and Reporting: The level of risk will be tracked, monitored, and reported throughout the trial lifecycle, implementing a TMF Risk Log to be updated on a regular basis by the trial team.
The TMF Risk Management Plan should be cross referenced by, or even be annexed to, the overall TMF Plan. Together, these documents form the foundation of your risk-based TMF framework.
TMF Risk Log
The TMF Risk Log serves as a dynamic tool for recording and tracking identified risks. It includes fields for capturing risk details, analysis outcomes, formulated responses, and current risk status. To be effective, the log must be regularly updated as new risks emerge or existing ones undergo changes. The log is a living document, crucial for maintaining transparency and facilitating effective risk management collaboration among team members.
Often, there is an overall trial risk assessment that is performed to identify all potential risks that could occur across all trial processes. This risk assessment can be used as the basis for the TMF Risk Log in addition to the standard risks that apply across all studies.
Step 2: assess Core vs Recommended documents
Because we are asked to develop and use a risk-based approach for performing TMF document QC and because not all TMF documents are essential to tell the story, a TMF document assessment must be done. This TMF document assessment helps to identify which documents carry the highest risks and may require more scrutiny in terms of QC or review.
In doing this kind of document assessment, different variables must be taken into consideration:
- If a document is core or recommended
- If the nature of the document has a direct effect on patient safety and/or data integrity
Core or Recommended
The first thing we need to evaluate is which documents are core and which ones are recommended. If a document is considered core, it means that it must be filed in the TMF, otherwise the inspector will not be able to reconstruct the story of the trial. This critical absence could have a negative impact on the final outcome of the inspection, or even the clinical trial itself. This evaluation should be performed and documented during the Master TMF Index customization.
It is crucial to understand which documents are necessary to reconstruct the story of the trial. Once this exercise has been performed on the Master TMF Index, we can then develop trial-specific templates or adapt the Master Index for a specific trial based on:
- The trial design (interventional vs non-intervention, device vs non-device, etc.)
- The operational model (outsourced vs insourced)
- Trial-specific SOPs (document output as process evidence)
- Geographic region-specific requirements (country-specific artifacts or requirements)
The CDISC TMF Reference Model identifies core and recommended documents, which is a good starting point. However, it should be noted that recommended documents may become core and vice versa based on the trial design.
When developing a plan for risk-based TMF oversight, you may leverage the core and recommended classifications to define different oversight strategies in conjunction with other risk evaluation factors.
Step 3: Evaluate impact on patient safety and data integrity
The next step is to perform an assessment on artifacts that could have an impact on patient safety and/or data integrity. We use the risk log developed in the first step to drive this exercise. For each artifact, an impact classification is assigned:

Some points to consider when performing an impact assessment include:
- This assessment could be done once for the organization in question and then applied across all clinical programs and studies.
- It should still be possible to make adjustments for specific study types or therapeutic areas if necessary.
- When identifying possible impacts, it is often useful to think about the processes that the artifacts play a role in.
- There could be multiple impacts for a specific artifact and therefore the most critical impact will determine the classification level assigned.
Step 4: Analyze non-compliance with TMF principles
The fourth assessment focuses on non-compliance with TMF principles (completeness, quality, and timeliness) which could impact risk. We should evaluate these principles and score them based on severity. We can apply a formula which provides an overall average risk score for a given artifact based on query rates, issue type and TMF dimensions (country, site, document owner, artifact type etc.).
The score is calculated as follows:
- We calculate a query rate risk score (QR) as outlined in table 1 for each issue type (I) per TMF dimension (D).
- We multiply each query rate risk score (QR) by the corresponding weighting (W) based on issue type (I) as outlined in Table 2 to calculate the weighted risk score (WR) for each issue type (I) and each dimension (D).
- We calculate the maximum weighted risk score (MR) by multiplying the highest query rate risk score (QR) by the highest weighting (W) for each issue type (I). In this example, it is always 12 as the highest query rate risk score is 4 and the highest weighting is 3.
- We then divide the weighted risk score (WR) by the maximum weighted risk score (MR) to obtain the dimension risk score % (DR) by dimension (D) and issue type (I).
- Finally we take each of the dimension risk scores (DR) and average them for the artifact in question to generate the artifact risk score (AR).
Query Rate Risk Score (QR)
When evaluating completeness, quality, and timeliness, the primary factor that we could leverage for risk assessment is query rate.
Query rate is defined as the % of artifacts that have valid queries raised on them. The higher the %, the higher the number of quality issues. Query rate can be calculated not only at the artifact level dimension (D) but also at the site, country, trial, organization, document owner, and process zone level dimensions. We assign a query rate risk score (QR) to each of these levels based on the % query rate, which is then used with a weighting coefficient (W) to determine the dimension risk score:
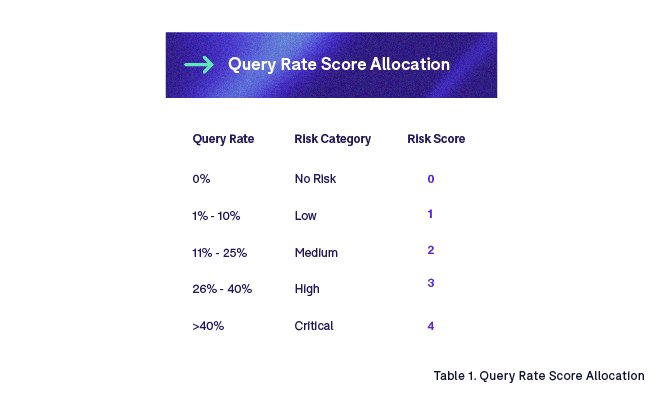
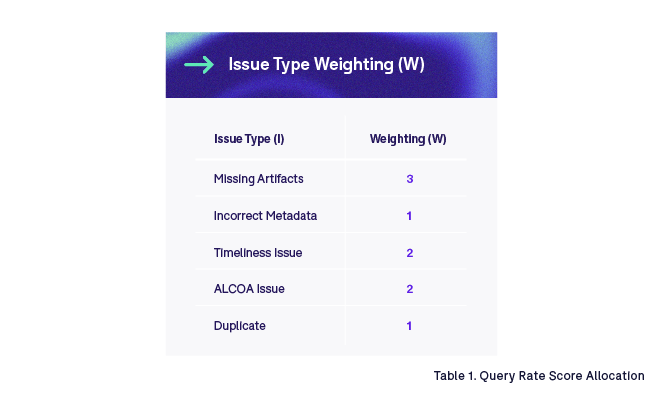
It may be of interest to further break down risk scores by issue type. For example, a missing artifact may present a higher risk than incorrect metadata. Proper evaluation should be performed to validate the need for such granularity. In our example, we apply a weighting to quality risk scores based on issue type. For instance:
Once we have defined the different query rate risk scores (QR) and weighting (W), we calculate the dimension risk score for each of the relevant dimensions for the artifact in question. The examples below show dimension risk scores for artifact and document owner levels for all monitoring visit reports (05.04.03) that are owned by John Doe.
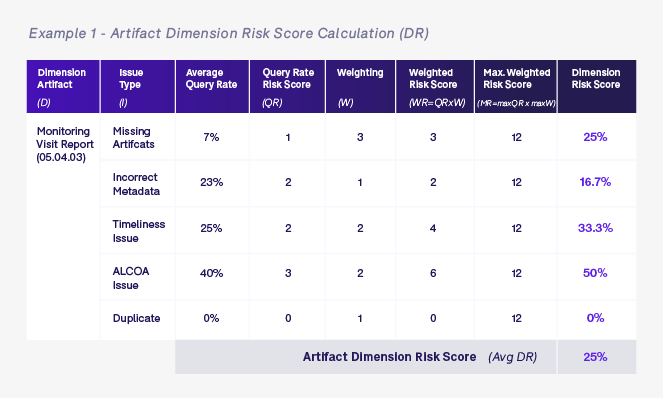
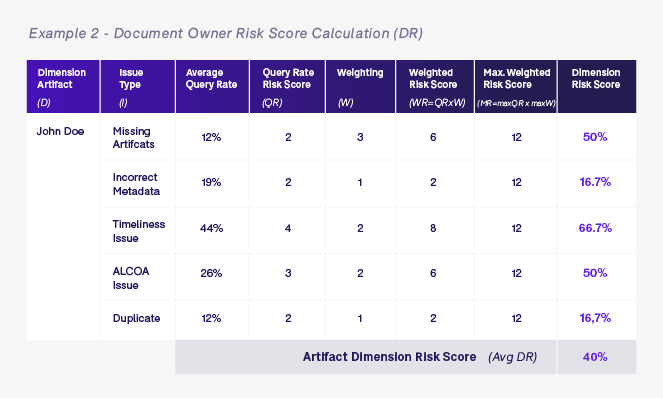
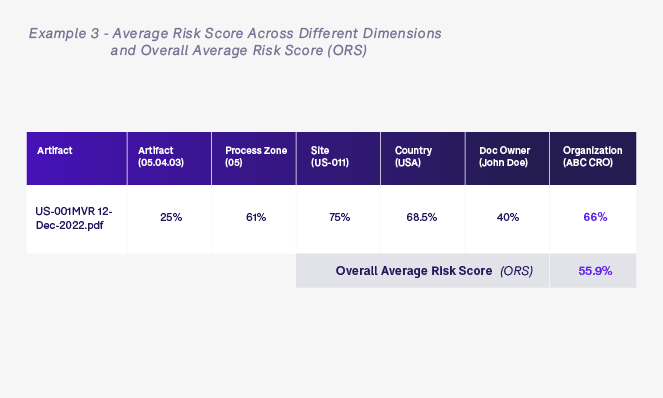
Once we have calculated all of the various dimension risk scores based on the artifact in question—in this case a monitoring visit report artifact 05.04.03 in process zone 05 for United States site US-001 that is owned by John Doe who works for ABC CRO—we calculate the overall average risk score (ORS). The ORS is simply the average of all dimension scores for this specific artifact and will be used to determine the level of oversight required for this artifact.
We may also want to take into consideration any queries that have been raised on this specific artifact in relation to quality and timeliness. We could add an additional risk score into the mix to take this into account.
Queries are typically raised when artifacts are being processed in the system either at the time of indexing, during initial QC, or during secondary QC and periodic review. In the future, we may see more queries being detected programmatically through the use of AI and edit checks.
We could also leverage historical query data to provide a larger body of data for our risk algorithms to provide even greater predictability.
Step 5: consider additional risk factors
Other risk factors may also be taken into account when calculating a risk score. These factors could include:
- Document Lifecycle: Does the document go through a workflow approval into the eTMF, or is it created outside the eTMF and uploaded as final? When the document goes through a document workflow inside the system, it means that it is drafted, reviewed and approved; because of this process, the quality is probably higher than a document created on a desktop and signed in wet ink from an ALCOA++ standpoint.
- Stand-Alone vs Package: Are there cross-checks to be done on this document? If the document is part of a larger group of artifacts required to describe a specific process or event, such as an ethics submissions and approval process following a protocol amendment, there could be many cross-checks that are required. On the contrary, if it is a stand-alone document such as an audit certificate, cross checks are unlikely to be required.
- Document Translation: Does the document need a formal translation? If so, we may need to do additional verification to ensure that the translation exists and is compliant.
- Document Location/Primary Source: eTMF is not the only system where TMF artifacts are stored—they can also be stored within other clinical systems with TMF signposts indicating where these records reside. These systems may not fall under the same TMF management team and therefore may represent additional risk from a TMF management standpoint.
- SOP Evidence: Most artifacts are governed by formal processes which are described in SOPs or trial plans. The procedural documents tend to dictate the documentation that must be produced, as well the content—and in some cases the format—of this documentation. In the case that an artifact is not governed by formal procedures, additional oversight may be required.
- Subject Recruitment Levels: Higher recruitment levels at clinical sites may require more oversight and present a higher risk of inspection.
- Key Trial Events: If specific key or higher-risk events occur, such as interim analysis, IP expiry extension, etc., then additional oversight may be required.
- Quality Events: Protocol deviations, procedural non-conformances, and any other trial-specific quality issues could increase risk factors.
- Notes to File: The existence of note to files may increase risk.
- Audit Findings: Any audit findings should also be evaluated and may increase risk scores.
- Trial Design Parameters: Certain aspects of trial design may increase risk, such as double-blind studies.
- Previous Performance: Evaluating previous performance in relation to completeness, quality, and timeliness for sites, countries, organizations and individuals could have an impact on risk scores.
These factors could generate additional scores which can be blended into the overall average risk score. Manually, it would be very laborious to use so many different factors; however, with technology it is possible to apply algorithms that will allow us to integrate these other factors.
As we develop more robust and technology-driven risk frameworks, and as data standardization and interoperability improves (i.e. DDF), we will be able to take into account a growing number factors to fine tune risk scoring. In the meantime, we need to find practical ways to highlight specific factors or events that allow us to adapt our RBA to consider the most important factors that can impact our ability to tell an accurate and complete story.
Using risk to optimize the quality check process
To optimize the quality check process, two distinct but impactful approaches can be followed: one focusing on a second level of risk-based QC with the assumption of a preliminary check on 100% of documents, and the other applying a risk-based approach at the first level of QC.
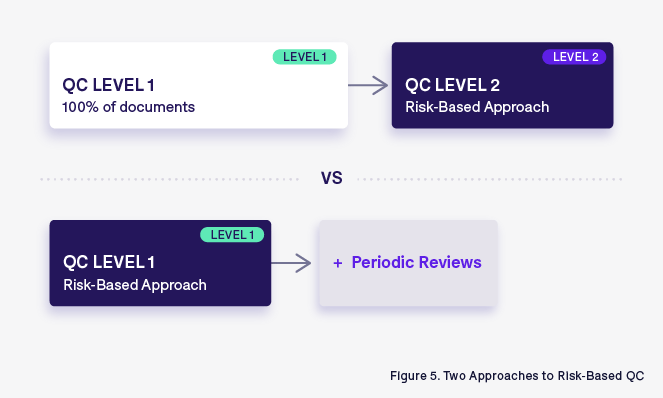
An RBA applied to the second level of QC:
Traditionally, the TMF quality check is composed of different levels of quality checks. The first level is done on every single document, with a focus of verifying compliance with ALCOA++ principles and GDocP. Once this exhaustive initial check is complete, a second level of QC, using RBA, is applied. This second level of QC is more focused on high-risk artifacts and aims to verify the links between TMF documents that enable the reconstruction of the entire story of the clinical trial.
While this approach can provide a comprehensive understanding of TMF inspection readiness, it is challenging—or sometimes impossible—to implement universally due to the large volume and diversity of checks that need to be undertaken. This is when a RBA can help us hone in on where the risks are highest or where issues may exist, and focus our secondary QC efforts in those areas.
The First level risk-based QC: Strategic and proactive
An alternative approach involves implementing a risk-based strategy at the first level of QC. Here, not every document undergoes the same level of scrutiny. Instead, documents are triaged based on their perceived risk. We consider factors such as criticality, impact on patient safety, and regulatory significance, and select artifacts that present the highest risks in these areas for QC rather than arbitrarily performing QC on 100% of all artifacts.
This approach takes into account the fact that not all artifacts are created equal, and that the QC process must be adapted to focus on higher-risk aspects of the trial.
When implementing such an approach, it is also important to perform periodic reviews to ensure that the risk-based approach is being applied adequately. One methodology for doing this would be to take a periodic sample of artifacts that were excluded from primary QC and perform a review.
If a significant number of errors are observed or artifacts were incorrectly excluded and present a higher level of risk than originally thought, then the risk-based approach should be adjusted.
Selecting the right approach
Choosing between these approaches requires careful consideration of trial specifics, timelines, and available resources. Some critical factors to consider include:
- Trial Complexity: High-risk trials may benefit from a more comprehensive initial QC.
- Resource Availability: Limited resources may steer teams towards a more targeted risk-based approach from the outset (first level of QC).
- Regulatory Landscape: Adherence to specific regulatory requirements may influence the chosen strategy.
- GDocP Knowledge: QC applied only to a portion of TMF documents can be more effective if we can ensure that document owners are trained and prepared on what ALCOA++ principles are.
- Lesson Learned: The experience with past TMFs can teach us that two levels of QC are better than one.
- Internal Processes: Company SOPs and WIs can be robust enough to support the approach of a second level of QC based on RBA.
- Quality Trends: Regular assessment of TMF quality issues can lead to a more valuable second-level RBA to QC (typing errors are not comparable to patient identifier errors).
How to leverage risk scores for TMF oversight
There are many paths that one can take to apply a risk-based approach. Using the methodology that we have outlined, we can then apply risk categorization and scores to define the amount of oversight that is necessary.
The first step is to evaluate the trial risk and impact on the TMF management process. The second step makes a distinction between core and recommended artifacts, and we define different oversight strategies for these two categories. The third step is to look at the risk classification in relation to an artifact’s impact on patient safety or data integrity. This is applied across studies. This classification combined with the average risk score calculated in step four and additional risk factors identified in step five will determine whether we only do an initial (ALCOA++) QC, or secondary QC and an additional periodic review. Finally, if historical data is available, we could also apply historical risk scores to specific artifacts to define whether a primary QC needs to be done at all. The two tables below outline sample risk-based oversight strategies for core and recommended documents.
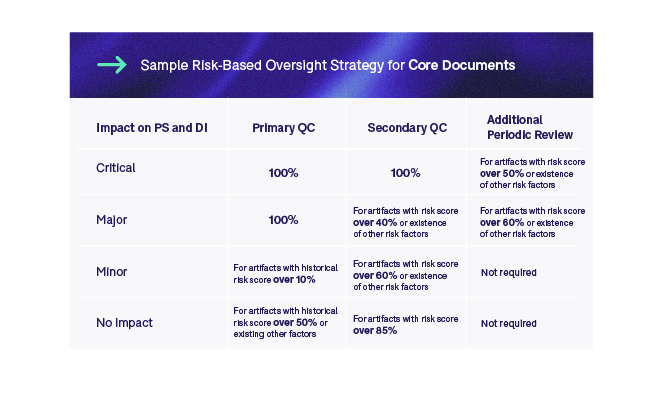
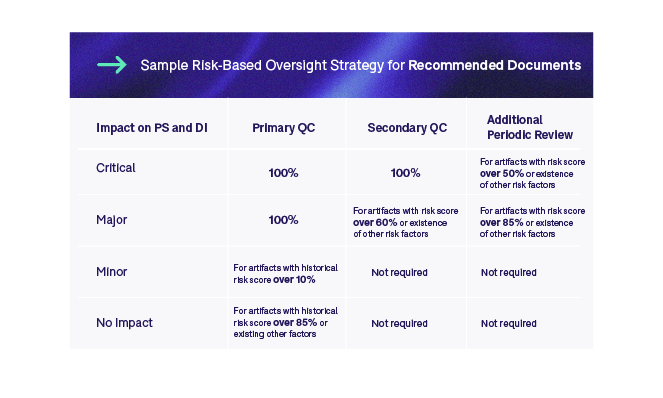
We could further introduce methodology whereby if we see an increase in error rate, we could then adjust the rules to increase the amount of oversight for specific artifact types. Whichever strategy we implement, it is imperative to properly validate it to ensure that it is adequate. Moreover, we must continuously monitor the outcomes of the RBA as the trial progresses and make adjustments where necessary.
Remember, not all studies are created equal, and therefore you will need to take a flexible approach to risk management within the TMF.
Conclusion
We have seen from the five-step approach outlined in this paper that taking a risk-based approach to TMF management and oversight is complex. It involves many different factors and large volumes of data and information that need to be analysed for our approach to be comprehensive. To perform this type of calculation manually is extremely challenging, which is why more simplistic, less comprehensive approaches have traditionally been taken—until now.
As eTMF solutions’ and clinical systems’ interoperability evolves and improves, we will start to be able to apply more robust approaches. Standardization of TMF-relevant information will also help in applying a system-based approach to risk management. Human oversight is and will remain important to ensure that the risk-based model that we implement is valid and remains so.
As we automate risk management more and more moving forward, we will be able to start better leveraging the value of our TMF management teams. These teams will be able to leverage the risk scoring information and risk management strategies to provide more focused, relevant oversight to our TMFs. In turn, this will have a positive impact on the completeness and quality of information contained within our TMFs, and our ability to tell a complete and accurate story of what occurred in a trial during inspections.
References
FDA (U.S. Food and Drug Administration):
FDA Guidance for Industry - "Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring" (August 2013).
FDA Guidance for Industry - "E8(R1) General Considerations for Clinical Studies" (November 2020).
FDA Guidance for Industry - "E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1)" (March 2018).
EMA (European Medicines Agency):
EMA Reflection Paper - "Risk-Based Quality Management in Clinical Trials" (November 2011).
EMA Guideline - "Good Clinical Practice: ICH E6(R2) Addendum" (November 2017).
MHRA (Medicines and Healthcare Products Regulatory Agency, UK):
MHRA Guidance - "Risk-adapted Approaches to the Management of Clinical Trials of Investigational Medicinal Products" (April 2011).
ICH (International Council for Harmonisation):
ICH Guideline - "E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1)" (November 2016).
WHO (World Health Organization):
WHO Guideline - "Handbook for Good Clinical Research Practice" (2016).
.jpg)


.jpg)
%20(and%20how%20to%20fix%20them)%20-%20Cover.png)
-1.png)
-1.png)

.png)


.png)